BlogData RoomsSalle de données virtuelle pour la biotechnologie 2026 : HIPAA, IND/NDA et essais cliniques
Salle de données virtuelle pour la biotechnologie 2026 : HIPAA, IND/NDA et essais cliniques
·14 min de lecture
Marc Seitz
Une salle de données virtuelle pour la biotechnologie est un espace de travail en ligne sécurisé utilisé pour gérer la documentation sensible que les entreprises de biotechnologie et des sciences de la vie partagent lors de levées de fonds, de licences, de fusions-acquisitions, d'essais cliniques et de dépôts réglementaires auprès de la FDA. Les VDR biotech nécessitent une conformité renforcée par rapport à la plupart des secteurs : HIPAA, FDA 21 CFR Part 11 et GCP (Bonnes Pratiques Cliniques) s'appliquent en plus des normes de base SOC 2, RGPD et ISO 27001. Papermark prend en charge les salles de données de niveau biotech à 99 €/mois forfaitaire avec un déploiement auto-hébergeable pour les charges de travail réglementées.
Récapitulatif rapide
Une salle de données virtuelle biotech est un référentiel en ligne sécurisé pour partager les dépôts IND/NDA, les données d'essais cliniques, les portefeuilles de brevets et les documents financiers avec les investisseurs, les partenaires de licence et les autorités réglementaires.
Conformité spécifique à la biotechnologie : HIPAA (données des patients), FDA 21 CFR Part 11 (dossiers et signatures électroniques), GCP (Bonnes Pratiques Cliniques) et directives ICH.
Catégories principales de documents : dépôts réglementaires IND et NDA, protocoles et données d'essais cliniques, portefeuille de brevets, correspondance avec la FDA, documentation de fabrication et informations financières.
Fonctionnalités essentielles pour la biotechnologie : filigrane dynamique, application de NDA, autorisations granulaires par partenaire, journal d'audit en ajout seul, résidence des données (UE/États-Unis) et option de déploiement auto-hébergé.
Cas d'usage courants de VDR biotech : levées de fonds Série A/B, négociations de licences, diligence raisonnable M&A, gestion documentaire des essais cliniques, réunions préalables à la soumission FDA.
Offre Papermark adaptée à la biotech : 99 €/mois forfaitaire avec déploiement auto-hébergeable, compatible HIPAA (avec BAA en entreprise), conformité RGPD et options de résidence des données en UE.
Qu'est-ce qu'une salle de données virtuelle pour la biotechnologie ?
Une salle de données virtuelle pour la biotechnologie est une plateforme en ligne sécurisée utilisée par les entreprises de biotechnologie, pharmaceutiques et des sciences de la vie pour stocker, organiser et partager des documents confidentiels avec les investisseurs, partenaires, régulateurs et sites d'essais cliniques. Elle fournit des autorisations granulaires, un filigranage dynamique, l'application de NDA et la piste d'audit requise pour la conformité à la FDA 21 CFR Part 11 et HIPAA.
La biotechnologie diffère des autres secteurs de deux manières importantes : les documents contiennent régulièrement des informations de santé protégées (PHI) ou des secrets commerciaux d'une valeur commerciale inestimable (propriété intellectuelle, données cliniques, procédés de fabrication), et la surveillance réglementaire est plus profonde que dans la plupart des secteurs. Une salle de données virtuelle pour la biotechnologie doit satisfaire au minimum à SOC 2 Type II et au RGPD, avec HIPAA et FDA 21 CFR Part 11 comme ajouts courants.
Pourquoi la biotechnologie a besoin d'une salle de données virtuelle spécialisée
Les entreprises de biotechnologie traitent des documents dont la fuite a de réelles conséquences juridiques, concurrentielles et réglementaires. Six raisons spécifiques poussent les entreprises de biotechnologie vers des salles de données virtuelles spécialisées.
Protection de la propriété intellectuelle. Les demandes de brevet, les secrets commerciaux, les données de recherche et les procédés de fabrication représentent la majeure partie de la valeur d'une entreprise de biotechnologie. Le filigranage dynamique et les autorisations granulaires préviennent les fuites qui détruiraient un futur dépôt de brevet.
Conformité réglementaire. La FDA 21 CFR Part 11 exige des dossiers électroniques avec des pistes d'audit, des signatures électroniques avec vérification d'identité et la validation du système. HIPAA exige un accès contrôlé aux PHI avec une couverture BAA pour tout fournisseur traitant ces données.
Négociations de licence. Les accords de licence en biotechnologie impliquent généralement de 3 à 10 partenaires potentiels, chacun avec ses propres équipes techniques et juridiques. Un accès délimité par partenaire et l'application des NDA sont essentiels.
Diligence pour les fusions-acquisitions. Les opérations de M&A dans le secteur biotechnologique impliquent d'importants ensembles de documents techniques (données d'essais cliniques, portefeuilles de brevets, dossiers de fabrication) partagés avec des acheteurs stratégiques et de capital-investissement.
Gestion des essais cliniques. Les essais multicentriques nécessitent un accès aux protocoles, aux formulaires de consentement éclairé, aux accords de site et aux données de sécurité pour les investigateurs, les moniteurs et les promoteurs.
Levée de fonds auprès d'investisseurs. Les levées de fonds de série A/B dans la biotechnologie impliquent une diligence scientifique et clinique détaillée, souvent avec des investisseurs qui font appel à leurs propres conseillers scientifiques dans la data room.
Documents spécifiques au secteur biotechnologique à inclure
Les data rooms biotechnologiques ajoutent 7 à 10 catégories à l'ensemble standard de documents pour les levées de fonds ou les opérations de M&A. Le tableau ci-dessous répertorie tous les documents biotechnologiques essentiels.
Document
Catégorie
Essentiel
Souhaitable
Mémorandum d'information confidentielle (CIM)
Présentation de l'entreprise
Résumé exécutif
Présentation de l'entreprise
Informations sur le comité consultatif scientifique
Présentation de l'entreprise
États financiers
Informations financières
Déclarations fiscales
Informations financières
Documents de constitution
Documents juridiques
Accords d'actionnaires
Documents juridiques
Demandes et approbations de brevets
Propriété intellectuelle
Accords de licence de PI (entrants et sortants)
Propriété intellectuelle
Politiques relatives aux secrets commerciaux
Propriété intellectuelle
Dépôts IND / NDA / BLA
Réglementaire
Correspondance avec la FDA
Réglementaire
Autorisations réglementaires (par juridiction)
Réglementaire
Protocoles d'essais cliniques
Recherche clinique
Données et résultats des essais cliniques
Recherche clinique
Formulaires de consentement éclairé
Recherche clinique
Brochures pour les investigateurs
Recherche clinique
Données de sécurité / Pharmacovigilance
Recherche clinique
Documentation des processus de fabrication
Fabrication
Procédures de contrôle qualité (BPF)
Fabrication
Projets de pipeline et feuille de route
Recherche et développement
Rapports de recherche et développement
Recherche et développement
Contrats clients et fournisseurs
Opérationnel
Contrats des employés et personnes clés
Ressources humaines
Partenariats et collaborations
Opérationnel
Analyse de marché
Marché et concurrence
Analyse concurrentielle
Marché et concurrence
Facteurs de risque
Complémentaire
Utilisation des fonds
Complémentaire
HIPAA, FDA 21 CFR Part 11 et GCP : la pile de conformité biotech
Les VDR biotech opèrent sous une pile de conformité plus approfondie que la plupart des secteurs. Quatre cadres réglementaires s'appliquent régulièrement.
HIPAA (Health Insurance Portability and Accountability Act). Requis dès que la salle de données contient des informations de santé protégées (PHI). Nécessite un accord de partenaire commercial (BAA) avec le fournisseur de VDR, la journalisation des audits, le chiffrement au repos et en transit, ainsi que des contrôles d'accès. Papermark prend en charge les déploiements conformes HIPAA via l'auto-hébergement et un BAA signé dans les plans entreprise.
FDA 21 CFR Part 11. Réglementation de la FDA américaine couvrant les dossiers électroniques et les signatures électroniques. Nécessite des pistes d'audit montrant qui a créé, modifié ou consulté les dossiers ; des signatures électroniques avec vérification d'identité ; une documentation de validation du système ; et des contrôles pour empêcher toute altération non autorisée des dossiers. Applicable aux dépôts IND, NDA, BLA et autres dépôts réglementaires connexes.
GCP (Good Clinical Practice). Lignes directrices ICH E6(R2) et directives connexes couvrant la conduite, la documentation et la surveillance des essais cliniques. Le VDR soutient les flux de travail GCP en maintenant des journaux d'audit immuables, des historiques de versions et un accès délimité pour les investigateurs, moniteurs et promoteurs.
RGPD et résidence des données régionale. Les essais menés dans l'UE nécessitent la conformité au RGPD et souvent la résidence des données dans l'UE. Les essais multi-juridictionnels nécessitent des contrôles d'accès par site respectant les lois locales sur la protection de la vie privée.
Comment configurer une salle de données biotech
Choisissez un VDR adapté à la biotech : exigez la prise en charge SOC 2 Type II, RGPD, HIPAA (avec BAA) et FDA 21 CFR Part 11. L'option d'auto-hébergement est importante pour les données cliniques sur site.
Construisez la structure de dossiers selon les catégories biotech standard : 1.0 Entreprise, 2.0 Financier, 3.0 Juridique, 4.0 PI, 5.0 Réglementaire (IND/NDA), 6.0 Recherche clinique, 7.0 Fabrication (cGMP), 8.0 R&D Pipeline, 9.0 Commercial.
Téléchargez les documents avec la convention de nommage YYYY-MM-DD_DocumentType_Indication.
Configurez les autorisations par groupe de partenaires ou d'investisseurs. Les prospects de licence et les soumissionnaires concurrents de fusions-acquisitions doivent être isolés.
Activez les fonctionnalités de sécurité : filigrane dynamique, protection par NDA, vérification par e-mail, restrictions de téléchargement et journalisation d'audit 21 CFR Part 11.
Testez la conformité avec votre responsable des affaires réglementaires avant d'ouvrir la salle aux évaluateurs externes.
Fonctionnalités de sécurité pour les salles de données virtuelles biotech
Contrôles de sécurité essentiels pour la biotech :
Chiffrement de bout en bout (AES-256 au repos, TLS 1.2+ en transit)
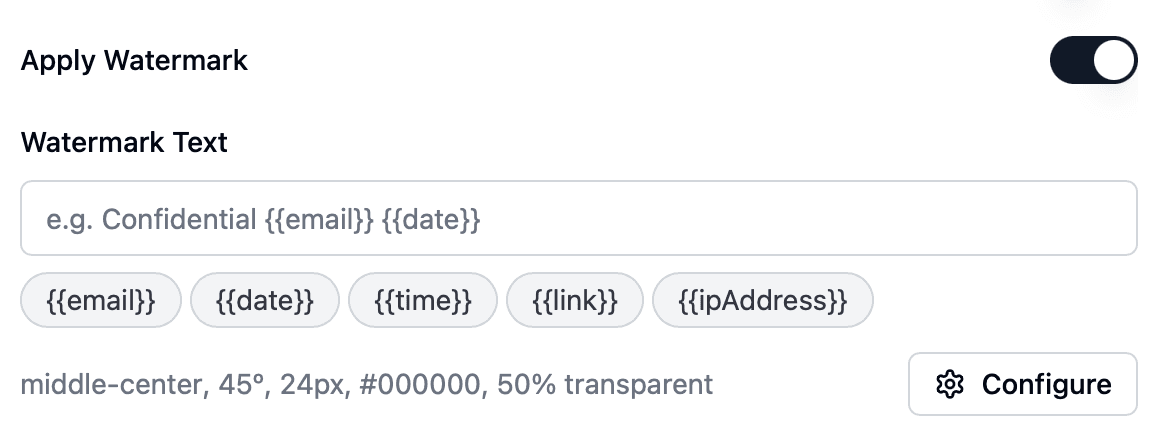
Filigrane dynamique par session avec e-mail du lecteur, IP, horodatage
Autorisations granulaires par partenaire, investisseur ou site clinique
Application de NDA avant l'accès aux documents
Vérification d'e-mail avec listes d'autorisation/blocage
Journal d'audit en ajout seul avec export pour revue FDA
Restrictions de téléchargement et de capture d'écran par lien
Expiration de lien liée aux délais de transaction ou de partenariat
Options de résidence des données (UE, États-Unis, régionales) pour les essais multi-juridictionnels
Étapes de transaction biotech et la salle de données requise
Les différentes étapes du cycle de vie d'une entreprise biotech nécessitent des configurations de salle de données distinctes. Le tableau ci-dessous met en correspondance les cinq étapes de transaction les plus courantes avec les documents et les modèles d'accès qu'elles requièrent.
Étape de transaction
Public principal
Catégories de documents clés
Modèle d'accès
Amorçage / Pré-IND
Investisseurs en capital-risque, family offices
R&D de pipeline, fondateurs scientifiques, dépôts de PI
Lecture seule avec filigrane, accès conditionné à NDA
Exigences HIPAA, FDA 21 CFR Part 11 et BPC associées aux fonctionnalités de la salle de données virtuelle
Chaque réglementation correspond à des contrôles techniques et procéduraux spécifiques. Le tableau ci-dessous établit cette correspondance de manière explicite.
Réglementation
Exigence
Fonctionnalité de la salle de données virtuelle
Règle de sécurité HIPAA
Contrôles d'accès
Accès basé sur les rôles, authentification multifacteur, dossiers à périmètre défini
Règle de sécurité HIPAA
Contrôles d'audit
Journal d'audit en ajout seul, conservation immuable
Règle de sécurité HIPAA
Intégrité
Contrôle de version des documents, vérification par hachage
Règle de sécurité HIPAA
Sécurité des transmissions
TLS 1.3, AES-256 au repos
Règle de confidentialité HIPAA
Minimum nécessaire
Autorisations à périmètre défini par destinataire
HIPAA / BAA
Accord d'associé commercial
BAA signé pour les forfaits entreprise
21 CFR Part 11
Signatures électroniques
Signature avec identité vérifiée et horodatage
21 CFR Part 11
Piste d'audit
Journal en ajout seul de tous les événements de création/modification/consultation
21 CFR Part 11
Validation du système
IQ/OQ/PQ documentés pour les déploiements auto-hébergés
21 CFR Part 11
Protection des enregistrements
Stockage de documents en lecture seule, historique des versions
BPC / ICH E6(R2)
Contrôle de version des documents
Historique des versions avec horodatages
BPC / ICH E6(R2)
Accès par site / par investigateur
Autorisations de dossier à périmètre défini
BPC / ICH E6(R2)
Accès du moniteur et du promoteur
Flux de révision pris en charge par le journal d'audit
RGPD (essais UE)
Résidence des données
Hébergement UE/Francfort, option auto-hébergée en UE
Erreurs courantes dans les data rooms biotech (et comment les éviter)
Cinq erreurs reviennent régulièrement dans les processus de diligence et de licence en biotechnologie.
1. Mélanger les documents contenant des PHI avec ceux qui n'en contiennent pas. Dès qu'un document de la data room contient des informations de santé protégées, l'ensemble de la data room doit respecter les contrôles HIPAA. Soit vous isolez les PHI dans une sous-salle dédiée conforme HIPAA, soit vous appliquez les contrôles HIPAA à l'ensemble de la data room.
2. Omettre le BAA avant de partager des PHI. L'accord de partenaire commercial (Business Associate Agreement) doit être signé avant l'arrivée des PHI dans la data room, et non rétroactivement. Vérifiez auprès du fournisseur de la plateforme.
3. Permettre aux investigateurs de sites cliniques de consulter les données d'autres sites. Les BPC exigent un cloisonnement par site. Un groupe d'autorisation global « tous les investigateurs » n'est pas conforme. Configurez des dossiers par site.
4. Traiter les IND/NDA comme des documents statiques. Les dépôts IND et NDA évoluent au fil des cycles de correspondance avec la FDA. Utilisez un système de contrôle des versions de documents et des dossiers horodatés, plutôt que de réécrire les dossiers.
5. Omettre la documentation de validation sur les déploiements auto-hébergés. Le 21 CFR Part 11 exige une qualification d'installation (IQ), une qualification opérationnelle (OQ) et une qualification de performance (PQ) pour tout système traitant des enregistrements réglementés. Documentez ces éléments avant la mise en production.
Cas d'usage spécifiques à la biotechnologie
Octroi de licences externes d'actifs de pipeline à des sociétés pharmaceutiques. Les sociétés biotech disposant d'un ou deux actifs en phase clinique s'associent souvent avec des laboratoires pharmaceutiques pour le développement avancé et la commercialisation. La data room héberge l'IND, le brochure d'investigateur, les résultats des essais cliniques, la documentation cGMP de fabrication, le portefeuille de propriété intellectuelle et la stratégie commerciale. Plusieurs équipes de développement commercial pharmaceutique peuvent évaluer en parallèle - les autorisations cloisonnées par soumissionnaire et le filigrane dynamique sont obligatoires.
Fusions-acquisitions de biotechs en phase clinique. Les acquéreurs stratégiques effectuent généralement 4 à 8 semaines de due diligence sur l'ensemble de la documentation d'entreprise, plus les éléments spécifiques à la biotech (pipeline réglementaire, données cliniques, fabrication). La data room doit prendre en charge 1 500 à 3 000 documents et 30 à 50 examinateurs sous la pression d'une transaction.
Diligence pour investisseurs série B/C et crossover. Les fonds sciences de la vie (RA Capital, ArrowMark, Perceptive, Cormorant) mènent une diligence institutionnelle incluant l'examen du pipeline, la validation du modèle financier, la liberté d'exploitation de la propriété intellectuelle et les vérifications de références de la direction. Le module Q&A avec réponses enregistrées préserve la piste d'audit pour les tours de financement suivants.
Essais initiés par l'investigateur. Les centres médicaux académiques et les biotechs co-gérant des essais initiés par l'investigateur nécessitent un accès délimité par site aux protocoles cliniques, aux FIC et aux rapports d'événements indésirables. Les pistes d'audit conformes aux BPC préservent la responsabilité réglementaire.
Soumissions de produits combinés (médicament + dispositif). Les soumissions à la FDA pour les produits combinés nécessitent à la fois le 21 CFR Part 11 (intégrité des données médicamenteuses) et la documentation QSR des dispositifs médicaux de la FDA. La data room doit prendre en charge les deux ensembles d'exigences de journaux d'audit.
Benchmarks des coûts VDR biotech
La tarification spécifique biotech tend à être plus élevée que la tarification VDR générale en raison de la charge de conformité. Fourchettes typiques 2026 :
Fournisseur
Niveau d'entrée (prêt biotech)
Notes
Papermark
99 €/mois forfaitaire (HIPAA via auto-hébergé + BAA entreprise)
Auto-hébergeable pour les exigences sur site clinique ou sponsor
Firmex
625 $/mois forfaitaire
Standard du secteur pour les salles de licences biotech
iDeals
Personnalisé (~10 000 $+/an)
Par page ou contrats annuels personnalisés
Datasite
Personnalisé (25 000 $+/an)
Courant pour les M&A biotech en phase avancée
Intralinks
Personnalisé (25 000 $+/an)
Courant pour les biotechs en cours d'introduction en bourse
Papermark prend en charge les déploiements biotech à 99 €/mois forfaitaire pour le plan Data Rooms, avec une version open source auto-hébergeable pour les charges de travail réglementées nécessitant un hébergement sur site :
Audité SOC 2 Type II
Conforme au RGPD avec options de résidence des données dans l'UE
Compatible HIPAA via déploiement auto-hébergé plus BAA signé (forfait entreprise)
Support 21 CFR Part 11 via déploiement auto-hébergé avec export des journaux d'audit
Filigrane dynamique, permissions granulaires, validation par NDA
Auto-hébergeable sur AWS, GCP ou infrastructure on-premise
Gérez le flux de transactions avec une data room virtuelle